The following chapter has been written by Andaleb Kholmukhamedov, M.D. and published in Platelets.
Abstract
There are two well-known subpopulations of activated platelets, pro-aggregatory and procoagulant. Procoagulant platelets represent a subpopulation of activated platelets, which are morphologically and functionally distinct from pro-aggregatory ones. Although various names have been used to describe these platelets in the literature (CoaT, CoaTed, highly activated, ballooned, capped, etc.), there is a consensus on their phenotypic features. Among them: exposure of high levels of phosphatidylserine (PSer) on the surface; decreased aggregatory and adhesive properties; support of active tenase and prothrombinase complexes; maximal generation by co-stimulation of glycoprotein VI (GPVI) and protease-activated receptors (PAR).
In this chapter, morphologic and functional features of procoagulant platelets, as well as the mechanisms of their formation, will be discussed.
Introduction
Blood has different components, like plasma, red blood cells (RBC), white blood cells (WBC), and platelets. Platelets, although being only “tiny fragments of megakaryocytes (mother cell),” are essential for life. We need them, together with about two dozen of coagulation factors, to keep all that nutrient-rich liquid plasma, infection-fighting WBCs and oxygen-carrying RBCs in our bodies in the case of trauma and bleeding. Platelets and coagulation get activated immediately upon being exposed to things they normally do not have contact with (e.g., during blood vessel wall rupture collagen gets exposed and activates platelets). Upon activation, platelets form a mesh-like structure using another plasma protein called fibrinogen as bridges between them. This process forms a so-called ‘unstable clot.’ At later stages of blood clotting, generated thrombin converts soluble fibrinogen to insoluble fibrin, stabilizing the initial platelet plug. These processes are collectively called hemostasis. Every aspect of hemostasis has its history. Although exploring history can be tedious, studying the bridge between past and present is essential in basic understanding of the subject, subject of procoagulant platelets in this case.
Unlike WBCs, which exist as functionally and morphologically distinct subpopulations, it has been thought for years that platelets are rather simple in their function, being just “cell particles.” It was later revealed that platelets, although lacking nuclear material, are indeed very complex not only in their nature but also in function. Nowadays, the existence of two different subpopulations of activated platelets, pro-aggregatory and procoagulant, is a widely accepted fact. Pro-aggregatory platelets, historically known as activated, have been a major focus since the initiation of platelet research. The history of procoagulant platelets, on the other hand, can be subdivided into two periods. The first one is the discovery of platelet procoagulant functionality, whereas in the second period, procoagulant platelets were discovered and characterized as a distinct subpopulation of activated platelets.
In 1912, long before we learned that prothrombinase supporting platelets are distinct from pro-aggregatory ones, Howell discovered that unsaturated cephalin is a phospholipid factor that triggers clotting (1). This discovery was followed by decades of controversial results of what phospholipid or what mixture of phospholipids is responsible for this effect until, in 1960, Karl Slotta demonstrated the presence of phosphatidylserine (PSer) is an absolute requirement (2). This discovery established the central role of PSer in prothrombinase activity, which in a way, paused research activities in the procoagulant platelet field.
The history was resumed when two decades later, Bevers and colleagues spectrophotometrically showed that thrombin and collagen co-stimulated platelets possess ~5 fold higher prothrombinase activity compared to those stimulated with thrombin or collagen alone. This effect was even more pronounced in the presence of exogenous factor Va (3). Later that year, the same group confirmed their spectrophotometric observations in one stage prothrombinase assay (4). In 1985, Rosing et al. more thoroughly described the role of platelet PSer exposure in prothrombin (FII) and factor X (FX) activation (5). They revealed that collagen and thrombin co-stimulation significantly increases prothrombinase and tenase activity, while this increase was abolished in the presence of phospholipase A2 (PLA2) from N. Naja. Also observing no cellular lysis in these conditions, they concluded that PSer exposure, which is followed by thrombin and collagen stimulation, is responsible for this phenomenon. In 1993, with the discoveries of annexin V and advancement in flow cytometry, Dachary-Prigent et al. established a protocol to detect PSer exposing platelets, the methodology that has overtime become fundamental in procoagulant platelet field and has been widely used since then (6). In 1997 Heemskerk and colleagues discovered that a percentage of platelets adhering to collagen, but not fibrinogen, balloon and expose PSer (7). In parallel to these findings, there was a series of publications determining the extent and ultrastructure of PSer exposure, as well as the essential role of calcium in this process (8–10).
Although numerous studies described platelet procoagulant function in response to dual stimulation (3,5,11,12), a breakthrough discovery that identified procoagulant platelets as a distinct subpopulation of activated platelets is the work by Alberio et al., where they demonstrated that only a certain percentage of activated platelets retain factor Va (FVa) on their surface (13). This subpopulation was generated by co-stimulation with convulxin/collagen (GPVI agonists) and thrombin; hence they were named CoaT platelets. A few years down the road, Kulkarni and Jackson introduced a new term – ‘sustained calcium-induced platelet morphology (SCIP)’ by discovering the fact that procoagulant platelets require prolonged elevation in cytosolic calcium to form (14). Diversifying terminology of procoagulant platelets did not stop there, in 2005 revealing that procoagulant platelets retain multiple α-granule proteins on their surface in transglutaminase dependent manner led to the introduction of a new term – ‘coated’ (by α-granule proteins) platelets (15,16). Further studies by Panteleev et al. revealed that this subpopulation of activated platelets binds high levels of factors IXa and Xa (17). In 2008, Jobe et al., characterizing molecular mechanisms of procoagulant platelet formation, introduced another term – highly activated platelets (18). There are few more terms used in the literature like ballooned (7), ballooned and procoagulant-spread (19) and super-activated platelets (20).
Just like in a famous Indian parable, where blind men try to describe an elephant they have never encountered before by touching different parts of it, researchers have been describing different (morphological and functional) features of procoagulant platelets and introducing different terms based on their discoveries. Whereas indeed, everyone has just been describing “different parts of the same elephant.”
Mechanisms of Procoagulant Platelet Formation
The discovery of procoagulant platelets as a distinct subpopulation of activated platelets at the beginning of the 21st century triggered research activities into cellular and molecular mechanisms of their formation. As shown by Alberio et al. and confirmed in later studies, procoagulant platelets are maximally generated upon co-stimulation of glycoprotein VI and PAR1/4. In 2005 Jobe and colleagues discovered that the absence of FcRγ, a key component responsible for glycoprotein VI signaling, ablates procoagulant platelet formation almost to baseline levels, evidencing GPVI stimulation is the major component of their generation (21). In the same year, Remenyi et al. demonstrated the role of mitochondrial permeability transition (MPT) in procoagulant platelet formation (16). MPT is a Ca2+-dependent molecular process that leads to mitochondrial swelling and cell death (22,23). During the onset of MPT, large pores are formed on the mitochondrial inner membrane making it non-specifically permeable to all solutes and molecules of molecular weight up to 1500 Da (24,25). It is a very well-known fact that mitochondrial Ca2+ overload can induce MPT, although the structure of MPT pore remains unknown.
Ca2+, being a key signaling molecule in most cells, is important for many processes, including platelet shape change and integrin α2bβ3 activation (26,27). In resting platelets, free Ca2+ is tightly regulated and maintained at about 100 nM in both the cytosol and mitochondria through the action of Ca2+-ATPases in the plasma and cell membranes. Thus, platelet cytosolic free Ca2+ is substantially lower than the blood Ca2+ levels, which are around 2 mM. With stimulation, however, cytosolic Ca2+ increases instantaneously. As outlined in Figure 1, this increase is mediated by activated phospholipase C (PLC). There are two major isoforms of PLC in human platelets, PLCβ and PLCγ. PLCβ is only activated downstream of Gq protein-coupled receptors (GPCRs). Whereas, PLCγ is activated downstream of numerous receptors like GPVI, glycoprotein Ib-IX complex (GPIb-IX), Fcγ receptor IIa (FcγRIIa), and C-type lectin-like receptor 2 (CLEC-2). As illustrated in Figure 1, both isoforms of PLC induce the release of Ca2+ from the dense tubular system (DTS) as well as activating transient receptor potential channel 3 (TRPC3). DTS release of Ca2+, in turn, triggers its extracellular entry through store-operated calcium entry (SOCE). For some time, it remained mysterious on how the release of DTS calcium stores into the cytosol induces more Ca2+ to flow into the cell, further increasing its cytosolic concentrations. However, with the discovery of core components of SOCE, everything falls into place. STIM1 and ORAI1 are parts of the same complex. When STIM1 senses drop in Ca2+ levels within the DTS, it signals to ORAI1 located on a cell membrane to allow Ca2+ passage from extracellular space into the cell. As evidenced by the study in the early 2000s, increased cytosolic Ca2+ levels are one of the requirements of procoagulant formation (14). Mitochondria, being the major and perhaps the only Ca2+ buffering system within platelets, equilibrate increased cytosolic Ca2+ following PAR and GPVI co-stimulation. The ensuing fate of mitochondrial Ca2+ overload is the opening of the mitochondrial permeability transition pore (MPTP), followed by PSer exposure and integrin deactivation (Figure 1), ultimately leading to a cell death. However, in the case of hemostasis, this cell death shall be considered physiologic since procoagulant platelets are essential.
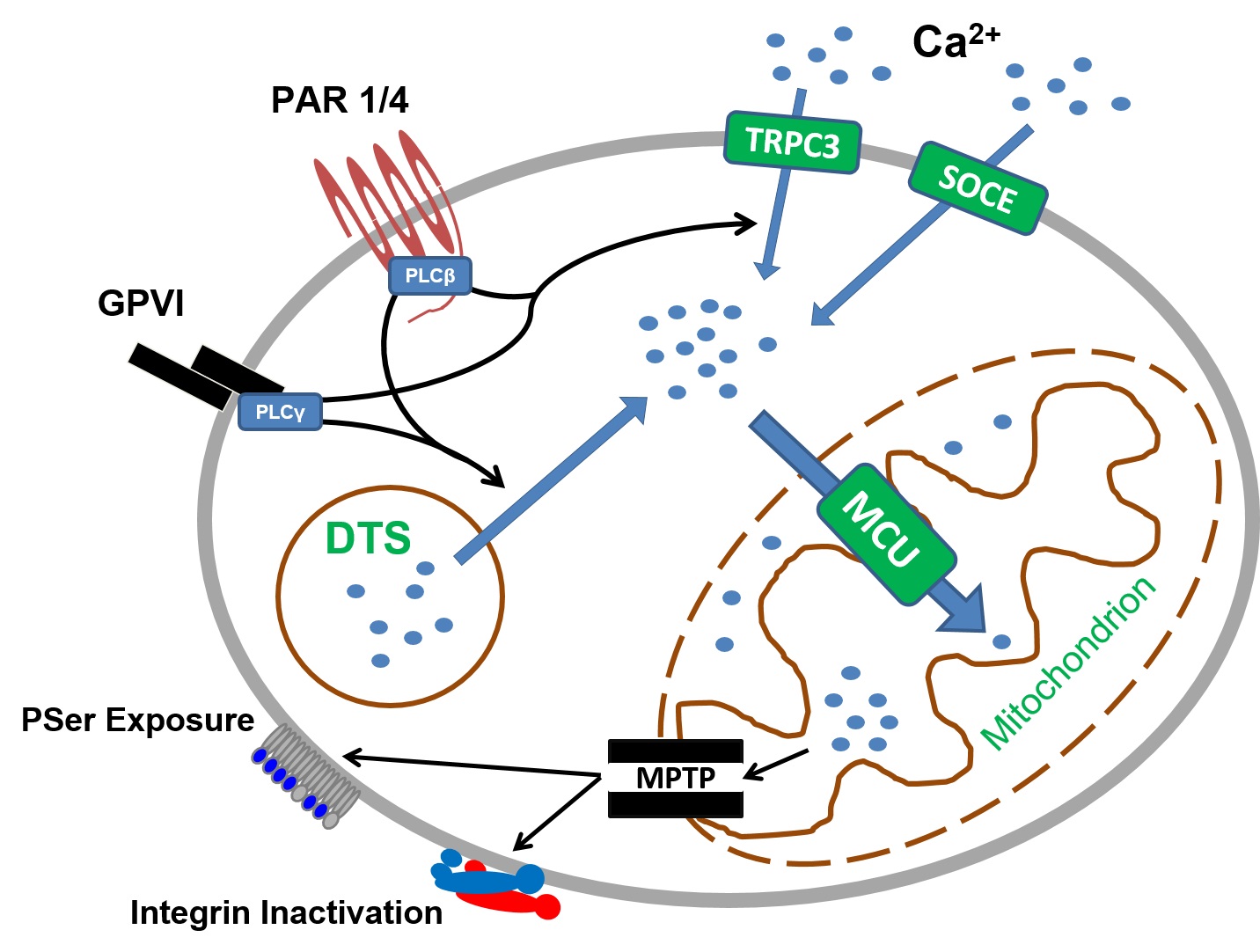
Figure 1. Molecular mechanisms of physiologic agonist-induced procoagulant platelet formation. In resting platelets low cytosolic Ca2+ is maintained by cell and plasma membrane Ca2+-ATP-ases. Co-stimulation of protease activated receptor 1/4 and glycoprotein VI leads to a sustained increase of cytosolic Ca2+ through activation of PLCβ and PLCγ, respectively. Increased cytosolic Ca2+ drives mitochondrial Ca2+ entry through the mitochondrial calcium uniporter complex. The resulting increase in mitochondrial Ca2+ opens mitochondrial permeability transition pore, which in turn leads to necrotic cell death and exposure of phosphatidylserine accompanied by integrin inactivation. DTS, dense tubular system; GPVI, glycoprotein VI; MCU, mitochondrial calcium uniporter; MPTP, mitochondrial permeability transition pore; PAR, protease activated receptor; PSer, phosphatidylserine; SOCE, store operated calcium entry; TRPC3, transient receptor potential channel 3. Adapted from (61) with modifications.
It brings us to another aspect of procoagulant platelet research, debated since their discovery, which is whether procoagulant platelets are necrotic or apoptotic. Cell death, both necrosis and apoptosis, is an essential event in the normal life of many cells in the human body, including platelets. Apoptosis, or programmed cell death, occurs in many organs throughout human lifetime. Necrosis, although considered to be mostly a catastrophic uncontrolled cell death, can also occur physiologically, as in the shedding of decidual endometrium during human menses. Necrosis and apoptosis differ in many aspects. The ultimate event during necrotic cell death is the osmotic swelling of the cell followed by the rupture of a cell membrane, whereas in apoptosis, cell shrinkage with preserved cell membrane is evident in later stages of this process.
Although the proposal that PSer is not homogeneously distributed on the surface of activated platelets was made back in 1985 (5), it took three decades to visualize that experimentally due to the complex nature of procoagulant platelets. In 2016 Podoplelova et al. presented a detailed structure of procoagulant platelets and cellular changes that give them the characteristic morphology. They elegantly demonstrated how the balloon is blown out from the platelet, leaving it as a “cap” (28). The existence of bulges in a phospholipid bilayer, known as the open canalicular system (OCS), is essential, as it provides platelet a fair amount of surface reserve for ballooning. Morphologic appearance, in this case, resemble the classical osmotic swelling of necrotic cell death, as schematically presented on the left side of Figure 2. Another feature of necrosis, which is a collapse of energy production, is present in procoagulant platelets, as they lack energy-requiring contractile function (18). Moreover, depletion of oxidative phosphorylation due to MPT, deactivates flippases that normally maintain the asymmetry of phospholipids. After the onset of MPT, contents of mitochondrial matrix get released to the cytosol leading to the second wave of cytosolic Ca2+ increase. This in turn leads to scramblase activation. Synergistically with deactivated flippases, scramblase performs its raison d’etre, which is flipping negatively charged PSer out in order to equilibrate its concentration. And finally, recent work demonstrated that platelets undergoing cell death through necrotic pattern and not apoptotic are functionally procoagulant as measured by prothrombinase support (29).
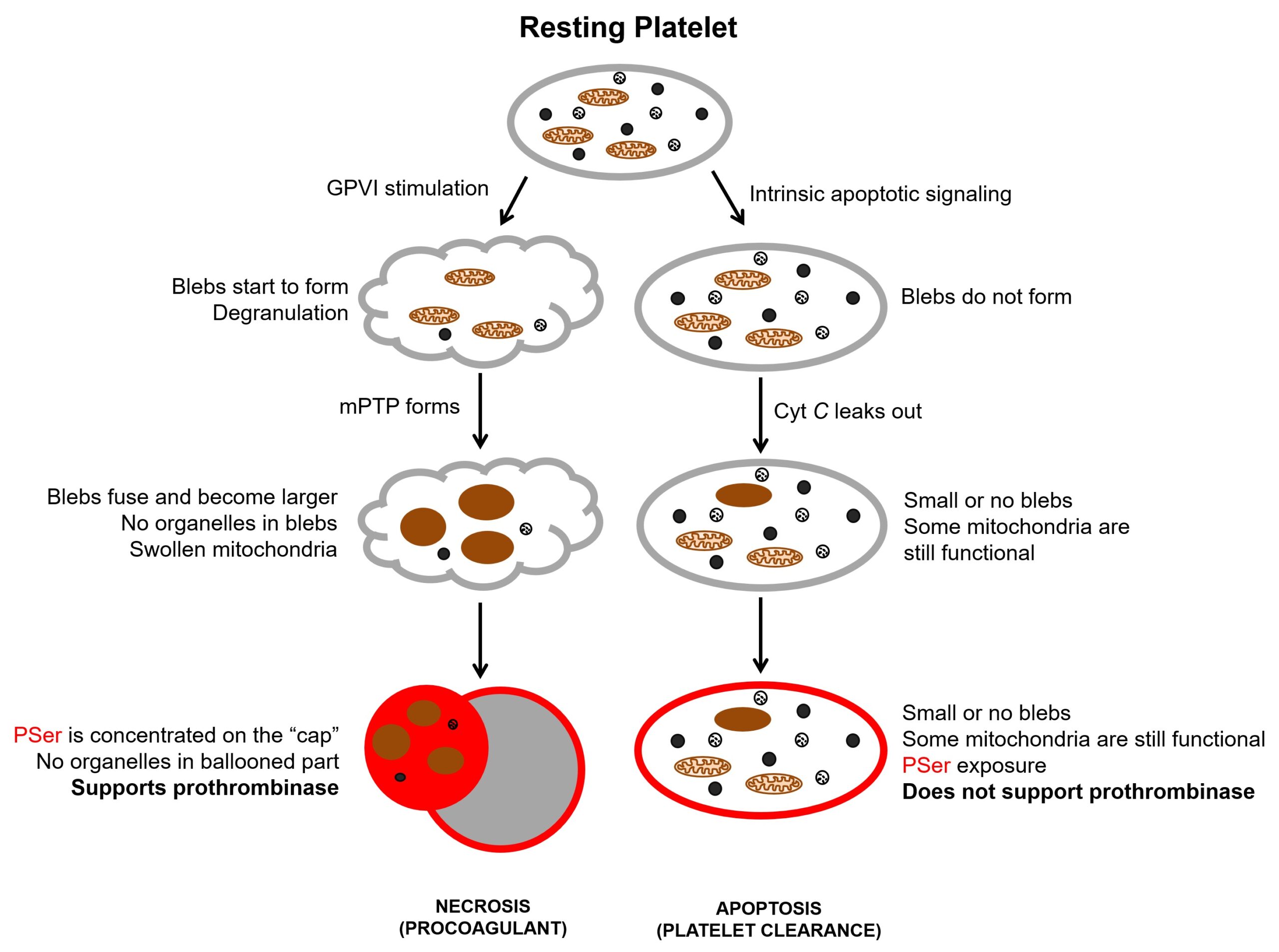
Figure 2. Cellular mechanisms of platelet cell death. Cyt C, cytochrome C; GPVI, glycoprotein VI; mPTP, mitochondrial permeability transition pore; PSer, phosphatidylserine. PSer exposure is shown in red.
Platelet apoptosis (Figure 2, right side), on the other hand, has been widely implicated in platelet lifespan via the action of the intrinsic mitochondrial apoptosis pathway (30,31). Normally, antiapoptotic members of the BCL-2 family (e.g., BCL-xL) restrain the activity of proapoptotic Bax and Bak proteins, which are present within the cytosolic fraction of a platelet. When BCL-xL wears off, oligomerized Bax and Bak translocate to the mitochondrial outer membrane and permeabilize it, which leads to cytochrome C leakage into the cytosol triggering apoptosome formation and eventually cell death. This is supported by the fact that the genetic ablation of murine BCL-xL reduces platelet life span from about five days to 5 hours (32,33).
Thus, procoagulant platelets are indeed necrotic, while apoptosis is essential in platelet clearance. It should, however, be mentioned that the presence of PSer on the surface of a platelet is indeed a signal for the reticuloendothelial system in the spleen and liver for clearance. Therefore, any procoagulant platelet that happened to escape the site of the active hemostatic process will be cleared out of the system by liver or spleen, just like an apoptotic platelet would.
Functions of Procoagulant Platelets
The physiological relevance of procoagulant platelets had been questioned for a long time. In recent years, however, after the demonstration of a procoagulant platelet being predictive of bleeding or ischemic complications in patients with coronary artery disease, brain hemorrhage, traumatic brain injury, stroke, etc. (34–41) it is gaining more and more recognition. The importance of this subpopulation is further highlighted on a novel ex vivo model (which integrates all the core components of hemostasis (42)), where pharmacologic inhibition of platelet transition to a procoagulant state without affecting pro-aggregatory phenotype results in a decreased thrombus stability (43).
It was initially thought that the only function of procoagulant platelets is to support coagulation. However, with recent advances in the field, we learn that depending on their localizations, procoagulant platelets can play different functions within the thrombus. For general consideration, these two functionalities will be discussed separately here.
Coagulation Support
Coagulation, a cascade of serine protease enzymatic reactions, is achieved by cleaving fibrinogen to fibrin, which transforms blood from a liquid to a gel-like state. Although platelet contribution to coagulation has been known for decades, the exact role of platelet phospholipids has been a matter of major debate. Dependence of hemostasis on biological membranes is very extensive, ranging from subendothelial membranes triggering coagulation and platelet activation to procoagulant platelet surface assembling tenase and prothrombinase complexes. It is very well known that at least two coagulation reactions are highly dependent on phospholipid surface, namely the activation of factor X and prothrombin by intrinsic tenase and prothrombinase, respectively. Both intrinsic tenase and prothrombinase are composed of the serine protease (FIXa for tenase and FXa for prothrombinase) and its protein cofactor (FVIIIa for tenase and FVa for prothrombinase). It is important to know that although both proteases alone are capable of activating their substrates, the presence of cofactors profoundly amplifies the catalytic process for up to 10,000 fold.
The current understanding of this process, based on numerous studies including mathematical modeling, is that phospholipid surface increases the rate of reactions by increasing the local concentration of coagulation factors, and thus increasing the probability of their interaction (28,44–50). This increase in a local concentration of factors, essential for tenase and prothrombinase complexes, is accomplished by the interaction of negatively-charged gamma-carboxyglutamic acid (GLA) residues of the coagulation factors with negatively-charged phosphatidylserine on the surface of procoagulant platelets. It is supported by the fact that GLA domain-containing (also known as vitamin K dependent) factors predominantly bind to a procoagulant subpopulation of activated platelets (17,28,29). Both enzymatic factors for intrinsic tenase (FIX) and prothrombinase (FX) are GLA domain-containing proteins and bind to PSer in a calcium-dependent manner. Whereas cofactors (FVIII and FV) structurally are not GLA proteins and bind to a procoagulant surface by different mechanisms.
In the case of FVIII, it has been shown by Gilbert et al. that it is not specific to a procoagulant surface (51). It was further confirmed by Podoplelova et al. in their efforts exploring procoagulant platelet characteristics, they demonstrated that both (pro-aggregatory and procoagulant) subpopulations of activated platelets bind FVIIIa (28). The fact that PS increases the catalytic activity of the intrinsic tenase complex by about 1500-fold (52) can be explained FIXa’s specificity to PSer. As outlined in Figure 3A, initially FIXa and FX bind PSer. This reaction is calcium-dependent for all GLA domain-containing proteins. FIXa possesses enzymatic activity to convert X to Xa, whereas in the presence of its cofactor (FVIIIa), the catalytic activity raises 100,000-fold.
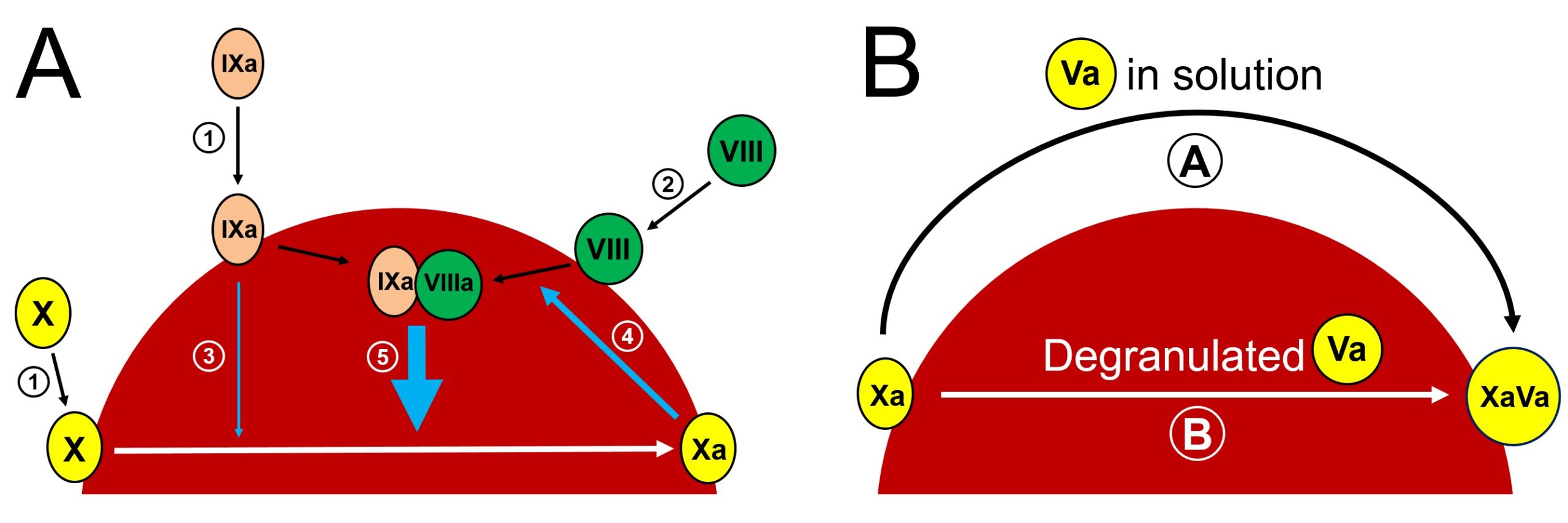
Figure 3. Factor X activation (A) and prothrombinase assembly (B) on the “cap” of procoagulant platelet. (A). Initially FIXa and FX are bound to PSer on the surface of procoagulant platelet in calcium dependent manner (①), whereas co-factor VIII binding is not spesific to PSer and does not require calcium (②). FIXa possesses enzymatic activity to convert X to Xa (③). Although reaction ③ is very slow in the absence of factor VIIIa, it is efficient to generate small amounts of FXa. Initial FXa then activates VIII to VIIIa (④), this leads to intrinsic tenase complex assembly, which in turn amplifies Xa formation (⑤). (B). There are two possibilities for the assembly of prothrombinase on the surface of procoagulant platelet. Pathway Ⓐ forms prothrombinase by the surface Xa reacting with factor Va in solution. Pathway Ⓑ depicts the possibility for prothrombinase complex formation by already bound Xa and Va. In this case both FVa and FXa form binary complexes with PSer first and then lipid-protein rearrangement leads to prothrombinase formation.
As for the FV – there are two different probabilities of binding to the procoagulant surface, as demonstrated in Figure 3B. In the first one, prothrombinase forms by the surface Xa reacting with factor Va in solution. Whereas, in the second case, both FVa and FXa form binary complexes with PSer first, and then lipid-protein rearrangement leads to prothrombinase formation (49). The addition of exogenous FVa to procoagulant platelets increases the velocity of prothrombinase reaction (4), whereas in the absence of exogenous FVa, procoagulant platelets are still prothrombinase active (53). These findings indicate that both pathways are physiologically important.
Limiting Thrombus Growth
During hemostasis, there is a time point when a thrombus needs to stop growing in order not to occlude the lumen of the vessel, which may compromise the blood supply to an end organ. Stoppage of the thrombus growth is probably the most intriguing part of thrombogenesis, although being the most understudied at the same time. When coagulation is initiated by subendothelial tissue factor, it leads to the formation of small amounts of thrombin. Thrombin then, via a positive feedback mechanism, by activating FXI and FVIII triggers the contact activation pathway and amplifies its production. This self-accelerating process is essential within the hemostatic plaque as it is the only way to overcome the anticoagulant nature of plasma, which is due to the presence of antithrombin III (ATIII). Besides the presence of ATIII in the active form, other mechanisms limit coagulation beyond its border zone. Another one of high importance is the presence of thrombomodulin, an integral membrane protein that is expressed ubiquitously on the surface of endothelial cells. Thrombomodulin converts procoagulant thrombin to an anticoagulant enzyme. Not only these mechanisms limit coagulation to the injured site but also degrade activated factors that happened to escape the hemostatic plaque. These processes outline the general principles of limiting coagulation to its border zone. However, not only coagulation but also processes of platelet adhesion, aggregation, and activation has to stop in order at a certain timepoint. How and when thrombus stops growing concerning cellular component had been a mystery for a long time.
In 2007 Maroney et al. demonstrated that procoagulant platelets express active tissue factor pathway inhibitor (TFPI) on their surface (54). But why platelets expressing procoagulant PSer would also need a strong anticoagulant (TFPI) on their surface remained a question for about a decade until in 2019 a breakthrough work by Nechipurenko et al. characterized a phenomenon, together with its mechanisms, of procoagulant platelets translocating to the thrombus periphery (55). They demonstrated that during clot retraction procoagulant platelets are squeezed out to the periphery of the thrombus, as shown in Figure 4. Another important study in this context is the 2008 work by Jobe et al., which demonstrated that procoagulant platelets do not possess contractile function (18). This explains why pro-aggregatory and procoagulant platelets have different translocation vectors within the hemostatic plaque (Figure 4). Being bound to the thrombus by its “cap” procoagulant platelets do not get detached but rather are squeezed out during pro-aggregatory platelets contraction. At the luminal surface of the hemostatic plaque procoagulant platelets limit its further growth not only by expressing low adhesive and aggregatory capabilities (56–58), but also with present TFPI, which would terminate any extrinsic tenase and prothrombinase activity.
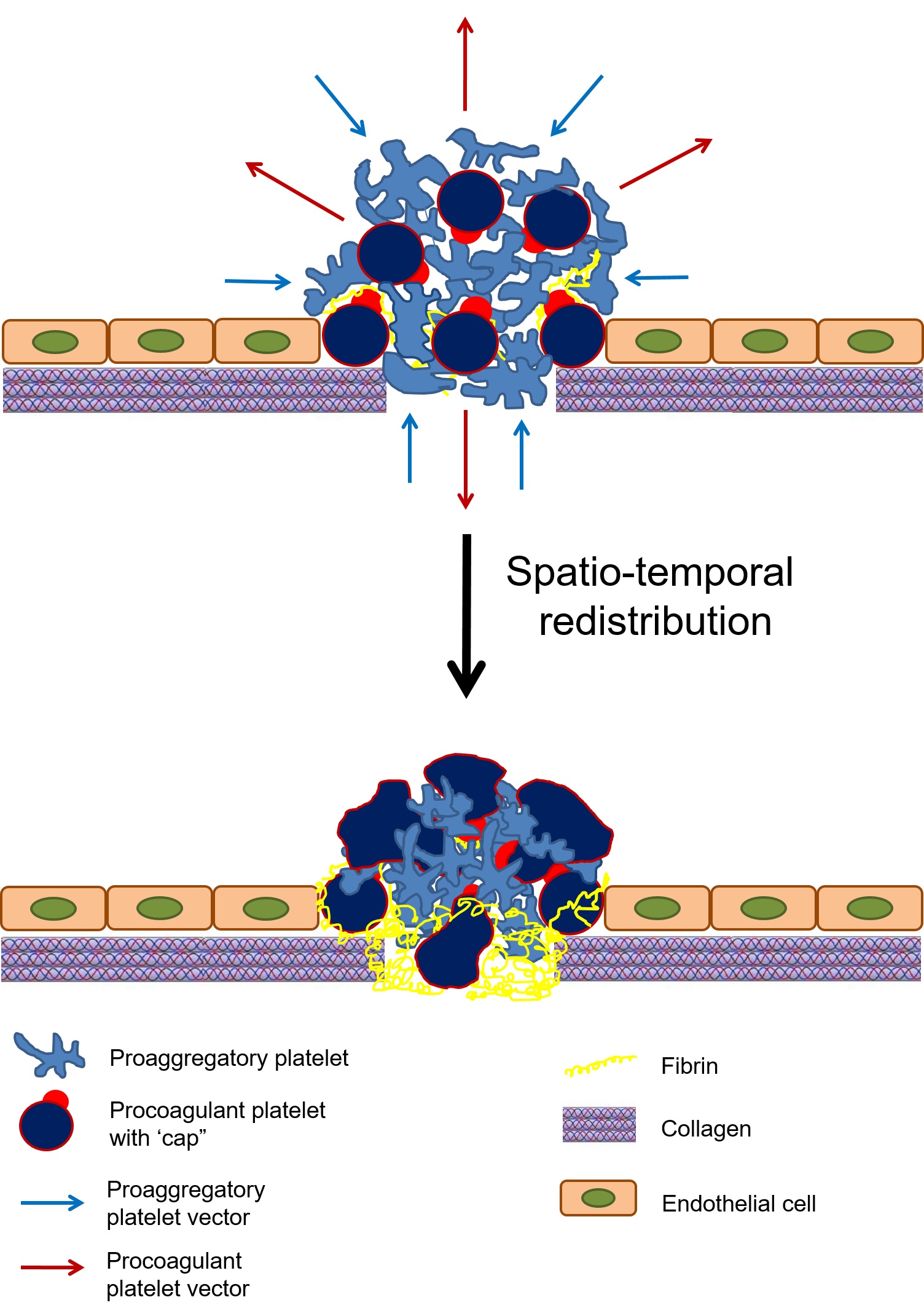
Figure 4. Schematic representation of platelet translocation within the evolving hemostatic plug. After vascular injury, platelets adhere and aggregate at the wound site. A subpopulation of platelets within the hemostatic plug transitions to a procoagulant state. Procoagulant and pro-aggregatory platelets have opposing vectors of translocation. Clot retraction, driven by platelets with a pro-aggregatory phenotype, squeezes procoagulant platelets to the periphery. Adapted from (62) with minor modification.
The fact of procoagulant platelet being non-adhesive, however, gives rise to a legitimate question. If procoagulant platelets are not capable of adhesion and aggregation, how do they get attracted to a thrombus? It turns out to be, as described in a recent studies, that procoagulant platelets do not form de novo from a resting state, but rather temporally transitioning from pro-aggregatory phenotype by the onset of the MPT and regulated necrosis within a hemostatic plaque (59,60).
Many are aware of a famous ancient Roman god of duality – Janus, who had two faces as he was able to look to the future and the past. Although not being as symmetrical as Janus, procoagulant platelets demonstrate duality by supporting coagulation on one side and limiting thrombus growth on the other.
References
- Howell W. Nature and Action of Thromboplastic (Zymoplastic) Substances of Tissues. Amer J Physiol. 1912;31:1–21.
- Slotta K. Thromboplastin. I. Phospholipid Moiety of Thromboplastin. PSEBM. 1960;(V103).
- Bevers EM, Comfurius P, van Rijn JL, Hemker HC, Zwaal RF. Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur J Biochem. 1982 Feb;122(2):429–36.
- Bevers EM, Comfurius P, Hemker HC, Zwaal RFA. On the Clot-Promoting Activity of Human Platelets in a One-Stage Prothrombinase Assay. Pathophysiol Haemost Thromb. 1982;12(3):268–74.
- Rosing J, van Rijn JL, Bevers EM, van Dieijen G, Comfurius P, Zwaal RF. The role of activated human platelets in prothrombin and factor X activation. Blood. 1985 Feb;65(2):319–32.
- Dachary-Prigent J, Freyssinet JM, Pasquet JM, Carron JC, Nurden AT. Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: a flow cytometry study showing a role for free sulfhydryl groups. Blood. 1993 May 15;81(10):2554–65.
- Heemskerk JW, Vuist WM, Feijge MA, Reutelingsperger CP, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood. 1997 Oct 1;90(7):2615–25.
- Trotter PJ, Orchard MA, Walker JH. Thrombin stimulates the intracellular relocation of annexin V in human platelets. Biochim Biophys Acta. 1994 Jun 30;1222(2):135–40.
- Trotter PJ, Orchard MA, Walker JH. Ca2+ concentration during binding determines the manner in which annexin V binds to membranes. Biochem J. 1995 Jun 1;308 ( Pt 2):591–8.
- Tzima E, Poujol C, Nurden P, Nurden AT, Orchard MA, Walker JH. Annexin V relocates to the periphery of activated platelets following thrombin activation: an ultrastructural immunohistochemical approach. Cell Biol Int. 1999;23(9):629–35.
- Smeets EF, Heemskerk JW, Comfurius P, Bevers EM, Zwaal RF. Thapsigargin amplifies the platelet procoagulant response caused by thrombin. Thromb Haemost. 1993 Dec 20;70(6):1024–9.
- Zwaal RF, Bevers EM. Platelet phospholipid asymmetry and its significance in hemostasis. Subcell Biochem. 1983;9:299–334.
- Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood. 2000 Mar 1;95(5):1694–702.
- Kulkarni S, Jackson SP. Platelet factor XIII and calpain negatively regulate integrin alphaIIbbeta3 adhesive function and thrombus growth. J Biol Chem. 2004 Jul 16;279(29):30697–706.
- Dale GL, Friese P, Batar P, Hamilton SF, Reed GL, Jackson KW, et al. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002 Jan 10;415(6868):175–9.
- Remenyi G, Szasz R, Friese P, Dale GL. Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscler Thromb Vasc Biol. 2005 Feb;25(2):467–71.
- Panteleev MA, Ananyeva NM, Greco NJ, Ataullakhanov FI, Saenko EL. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J Thromb Haemost JTH. 2005 Nov;3(11):2545–53.
- Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR, et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood. 2008 Feb 1;111(3):1257–65.
- Agbani EO, van den Bosch MTJ, Brown E, Williams CM, Mattheij NJA, Cosemans JMEM, et al. Coordinated Membrane Ballooning and Procoagulant Spreading in Human Platelets. Circulation. 2015 Oct 13;132(15):1414–24.
- Mazepa M, Hoffman M, Monroe D. Superactivated platelets: thrombus regulators, thrombin generators, and potential clinical targets. Arterioscler Thromb Vasc Biol. 2013 Aug;33(8):1747–52.
- Jobe SM, Leo L, Eastvold JS, Dickneite G, Ratliff TL, Lentz SR, et al. Role of FcRγ and factor XIIIA in coated platelet formation. Blood. 2005 Dec 15;106(13):4146–51.
- Choo H-J, Kholmukhamedov A, Zhou C, Jobe S. Inner mitochondrial membrane disruption links apoptotic and agonist-initiated phosphatidylserine externalization in platelets. Arterioscler Thromb Vasc Biol. 2017 Aug;37(8):1503–12.
- Kim J-S, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003 May 9;304(3):463–70.
- Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976 Aug 25;251(16):5069–77.
- Bernardi P, Forte M. The mitochondrial permeability transition pore. Novartis Found Symp. 2007;287:157–64; discussion 164-169.
- Hathaway DR, Adelstein RS. Human platelet myosin light chain kinase requires the calcium-binding protein calmodulin for activity. Proc Natl Acad Sci U S A. 1979 Apr;76(4):1653–7.
- Shattil SJ, Brass LF. Induction of the fibrinogen receptor on human platelets by intracellular mediators. J Biol Chem. 1987 Jan 25;262(3):992–1000.
- Podoplelova NA, Sveshnikova AN, Kotova YN, Eckly A, Receveur N, Nechipurenko DY, et al. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood. 2016 29;128(13):1745–55.
- Kholmukhamedov A, Jobe S. Necrotic but Not Apoptotic Platelets Are Functionally Procoagulant. Blood; 2018. 132 Suppl 1. p. 2420.
- McArthur K, Chappaz S, Kile BT. Apoptosis in megakaryocytes and platelets: the life and death of a lineage. Blood. 2018 08;131(6):605–10.
- Pleines I, Lebois M, Gangatirkar P, Au AE, Lane RM, Henley KJ, et al. Intrinsic apoptosis circumvents the functional decline of circulating platelets but does not cause the storage lesion. Blood. 2018 12;132(2):197–209.
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007 Mar 23;128(6):1173–86.
- Josefsson EC, James C, Henley KJ, Debrincat MA, Rogers KL, Dowling MR, et al. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med. 2011 Sep 26;208(10):2017–31.
- Prodan CI, Vincent AS, Dale GL. Coated-Platelet Levels Increase with Number of Injuries in Patients with Mild Traumatic Brain Injury. J Neurotrauma. 2016 01;33(9):818–24.
- Prodan CI, Stoner JA, Dale GL. Acute hemorrhagic complications are associated with lower coated-platelet levels in non-lacunar brain infarction. J Thromb Haemost JTH. 2015 Dec;13(12):2233–9.
- Prodan CI, Stoner JA, Dale GL. Lower Coated-Platelet Levels Are Associated With Increased Mortality After Spontaneous Intracerebral Hemorrhage. Stroke. 2015 Jul;46(7):1819–25.
- Prodan CI, Vincent AS, Dale GL. Coated-platelet levels are persistently elevated in patients with mild traumatic brain injury. J Head Trauma Rehabil. 2014 Dec;29(6):522–6.
- Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Coated-platelets predict stroke at 30 days following TIA. Neurology. 2017 Jul 11;89(2):125–8.
- Kirkpatrick AC, Stoner JA, Dale GL, Rabadi M, Prodan CI. Higher Coated-Platelet Levels in Acute Stroke are Associated with Lower Cognitive Scores at Three Months Post Infarction. J Stroke Cerebrovasc Dis Off J Natl Stroke Assoc. 2019 Sep;28(9):2398–406.
- Kirkpatrick AC, Tafur AJ, Vincent AS, Dale GL, Prodan CI. Coated-platelets improve prediction of stroke and transient ischemic attack in asymptomatic internal carotid artery stenosis. Stroke. 2014 Oct;45(10):2995–3001.
- Ray B, Pandav VM, Mathews EA, Thompson DM, Ford L, Yearout LK, et al. Coated-Platelet Trends Predict Short-Term Clinical OutcomeAfter Subarachnoid Hemorrhage. Transl Stroke Res. 2018;9(5):459–70.
- Sakurai Y, Hardy ET, Ahn B, Tran R, Fay ME, Ciciliano JC, et al. A microengineered vascularized bleeding model that integrates the principal components of hemostasis. Nat Commun. 2018 06;9(1):509.
- Kholmukhamedov A, Jobe S. Mitochondrial Uncoupling Prevents Procoagulant Platelet Formation Resulting in Decreased Thrombus Stability. Blood; 2018. 132 Suppl 1. p. 2421.
- Panteleev MA, Saenko EL, Ananyeva NM, Ataullakhanov FI. Kinetics of Factor X activation by the membrane-bound complex of Factor IXa and Factor VIIIa. Biochem J. 2004 Aug 1;381(Pt 3):779–94.
- Ataullakhanov FI, Panteleev MA. Mathematical modeling and computer simulation in blood coagulation. Pathophysiol Haemost Thromb. 2005;34(2–3):60–70.
- Panteleev MA, Balandina AN, Lipets EN, Ovanesov MV, Ataullakhanov FI. Task-Oriented Modular Decomposition of Biological Networks: Trigger Mechanism in Blood Coagulation. Biophys J. 2010 May 5;98(9):1751–61.
- Ovanesov MV, Ananyeva NM, Panteleev MA, Ataullakhanov FI, Saenko EL. Initiation and propagation of coagulation from tissue factor-bearing cell monolayers to plasma: initiator cells do not regulate spatial growth rate. J Thromb Haemost JTH. 2005 Feb;3(2):321–31.
- Sinauridze EI, Kireev DA, Popenko NY, Pichugin AV, Panteleev MA, Krymskaya OV, et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost. 2007 Mar;97(3):425–34.
- Krishnaswamy S, Nesheim ME, Pryzdial EL, Mann KG. Assembly of prothrombinase complex. Methods Enzymol. 1993;222:260–80.
- Mann KG, Krishnaswamy S, Lawson JH. Surface-dependent hemostasis. Semin Hematol. 1992 Jul;29(3):213–26.
- Gilbert GE, Novakovic VA, Shi J, Rasmussen J, Pipe SW. Platelet binding sites for factor VIII in relation to fibrin and phosphatidylserine. Blood. 2015 Sep 3;126(10):1237–44.
- Gilbert GE, Arena AA. Activation of the factor VIIIa-factor IXa enzyme complex of blood coagulation by membranes containing phosphatidyl-L-serine. J Biol Chem. 1996 May 10;271(19):11120–5.
- Nesheim ME, Nichols WL, Cole TL, Houston JG, Schenk RB, Mann KG, et al. Isolation and study of an acquired inhibitor of human coagulation factor V. J Clin Invest. 1986 Feb;77(2):405–15.
- Maroney SA, Haberichter SL, Friese P, Collins ML, Ferrel JP, Dale GL, et al. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007 Mar 1;109(5):1931–7.
- Nechipurenko DY, Receveur N, Yakimenko AO, Shepelyuk TO, Yakusheva AA, Kerimov RR, et al. Clot Contraction Drives the Translocation of Procoagulant Platelets to Thrombus Surface. Arterioscler Thromb Vasc Biol. 2019;39(1):37–47.
- Mattheij NJA, Gilio K, van Kruchten R, Jobe SM, Wieschhaus AJ, Chishti AH, et al. Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J Biol Chem. 2013 May 10;288(19):13325–36.
- Liu F, Gamez G, Myers DR, Clemmons W, Lam WA, Jobe SM. Mitochondrially mediated integrin αIIbβ3 protein inactivation limits thrombus growth. J Biol Chem. 2013 Oct 18;288(42):30672–81.
- Yakimenko AO, Verholomova FY, Kotova YN, Ataullakhanov FI, Panteleev MA. Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys J. 2012 May 16;102(10):2261–9.
- Alberio L, Ravanat C, Hechler B, Mangin PH, Lanza F, Gachet C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb Haemost. 2017 02;117(6):1101–14.
- Kholmukhamedov A, Jobe S. Mitochondria and Platelet Cell Death. Thromb Haemost. 2017;117(11):2207–8.
- Kholmukhamedov A, Janecke R, Choo H-J, Jobe SM. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J Thromb Haemost JTH. 2018;16(11):2315–21.
- Kholmukhamedov A, Jobe S. Procoagulant Platelets Get Squeezed to Define the Boundaries of the Hemostatic Plug. Arterioscler Thromb Vasc Biol. 2019;39(1):5–6.